Encefalopatiile spongiforme (boli prionice) sunt acele boli în care formele patologice ale proteinelor prionice sunt implicate în dezvoltare. Știm din ce în ce mai multe despre bolile prionice, dar aspectele cheie sunt încă necunoscute - în prezent medicina nu are mijloacele de a vindeca pacienții de aceste boli.
Encefalopatiile spongiforme sau bolile prionice se pot dezvolta în timpul vieții, în timp ce altele apar din mutații genetice moștenite prezente de la naștere. În cadrul acestui grup, există mai multe entități care apar la oameni, exemple sunt boala Creutzfeldt-Jakob sau insomnia familială fatală.
Bolile prionice au fost mult timp foarte misterioase. Spre deosebire de alți agenți patogeni, cum ar fi bacteriile, virusurile sau ciupercile, acestea nu conțin acid nucleic - prionii sunt compuși doar din proteine. Teoria bolilor prionice a fost descoperită de S. Prusiner, această descoperire a fost foarte apreciată în comunitatea științifică - în 1997 cercetătorul a primit Premiul Nobel pentru medicină. Deși au trecut relativ mulți ani de când s-a născut conceptul prion, unii oameni de știință încă cred că este incomplet și investighează în continuare natura acestor afecțiuni - unii dintre factorii responsabili pentru encefalopatiile spongiforme au fost acum confirmați.
$config[ads_text1] not found
Bolile prionice: cauze
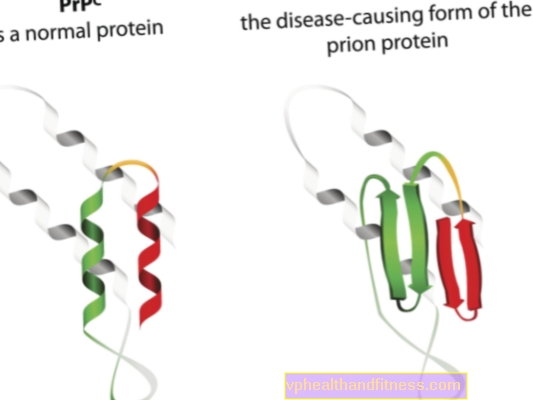
Etiologia bolilor prionice este legată de transformarea proteinelor prionice normale în forme patogene, patogene. Prionii sunt molecule de proteine care se găsesc în corpul fiecărei ființe umane. Funcția lor nu este încă complet clară, dar se știe că, în condiții normale, proteinele prionice nu dăunează organismului. Cu toate acestea, atunci când prionii își schimbă structura și devin particule patogene, se dezvoltă una dintre mai multe encefalopatii spongiforme. Prionii care apar în mod natural în organism sunt denumiți PRPC, în timp ce formele anormale sunt denumite PRPSC. Acestea din urmă reprezintă o problemă gravă nu numai pentru că se pot acumula în țesutul nervos sub formă de depozite și îi pot produce daune, ci și pentru că au capacitatea de a transforma prionii normali într-o formă malformată (pur și simplu, PRPSC poate „infecta” proteinele normale cu potențialul său patogen).
Citește și: Boala Huntington (coreea Huntington): cauze, simptome, tratament Fremături musculare - cauze. Ce înseamnă tremor muscular? Boli care ucid cel mai rapid: ȘOC, EBOLA, DAMN, ATAC, URGENȚĂ [GALE ...Practic, există 3 cauze ale encefalopatiilor spongiforme:
$config[ads_text2] not found- sporadică (mutația patogenă apare în celulele somatice, apare în timpul vieții pacientului),
- familie (rezultată din povara mutațiilor moștenite de la părinți),
- trecute (legate de introducerea prionilor patogeni în corpul uman, de exemplu, prin preparate cu hormoni de creștere contaminate cu aceste particule sau transplant de cornee de la o persoană care suferă de o anumită encefalopatie spongiformă).
Encefalopatii spongiforme: boala Creutzfeldt-Jakob
Boala Creutzfeldt-Jakob (CJD) a fost descrisă pentru prima dată la începutul anilor 1920. Există 4 tipuri de boală:
- CJD sporadică (cea mai frecventă, reprezentând până la 9/10 din toate cazurile de CJD)
- orașul natal al CJD
- CJD centurat
- varianta CJD
Tabloul clinic în cursul diferitelor variante ale bolii Creutzfeldt-Jakob poate fi variabil. Cele mai frecvente afecțiuni în cursul acestui grup de encefalopatii spongiforme sunt:
- tulburări de demență (inclusiv deteriorarea progresivă a memoriei, atenție și concentrare)
- mioclonie (mișcări involuntare, cum ar fi mișcări bruște ale mușchilor)
- disfuncție cerebelară (manifestată, de exemplu, prin tulburări de echilibru)
- vedere încețoșată
- simptome piramidale și extrapiramidale
În cursul variantelor CJD, pot apărea, de asemenea, tulburări mentale (de ex. Anxietate, dispoziție depresivă), durere și alte mișcări involuntare, altele decât cele menționate mai sus.
$config[ads_text3] not found
Prognosticul pentru boala Creutzfeldt-Jakob este slab - de exemplu, la pacienții cu BCJ sporadică, durează în medie patru până la cinci luni de la apariția simptomelor bolii până la moarte.
Encefalopatii spongiforme: sindromul Gerstmann-Straussler-Scheinker
Sindromul Gerstmann-Straussler-Scheinker (GSS) se desfășoară de obicei în familii și este cauzat de o mutație moștenită a genei PRNP. Este considerată a fi cea mai lentă progresie a encefalopatiei spongiforme. Echipa GSS include:
- ataxie spinocerebeloasă
- disartrie
- tulburări de demență
- tulburări de deglutiție
- nistagmus
- tensiune musculară crescută
Pacienții diagnosticați cu GSS au o perioadă de timp variabilă, iar la unii pacienți moartea are loc la mai mult de 10 ani de la debut.
Encefalopatii spongiforme: insomnie familială fatală
Insomnia familială letală este o boală prionică cauzată de mutații ale genei PRNP. Boala este extrem de rară și până acum a fost diagnosticată la 28 de familii din întreaga lume. În cursul insomniei familiale fatale, primul simptom este incapacitatea de a dormi. Această problemă are ca rezultat tulburări de anxietate și pacientul se confruntă cu halucinații. Efectul lipsei constante de odihnă nocturnă sunt tulburări în funcționarea sistemului autonom (inclusiv modificări ale funcției inimii, transpirații și tulburări ale sistemului digestiv), există, de asemenea, o scădere progresivă a greutății corporale. În stadiile mai avansate de insomnie familială fatală, apar tulburări hormonale, iar simptomele demenței apar în cursul bolii.
Prognosticul pentru insomnie familială fatală, ca și pentru alte encefalopatii spongiforme, este slab: pacienții mor de obicei în decurs de trei ani de la debut.
Encefalopatii spongiforme: prionopatie cu susceptibilitate variabilă la protează
Apariția acestor encefalopatii spongiforme este legată în principal de mutații ale genei PRNP. Cu toate acestea, aceste mutații privesc codoni diferiți ai acestei gene și, prin urmare, se disting mai multe boli prionice diferite. O unitate relativ recent descrisă (în 2008) este prionopatia cu susceptibilitate variabilă la protează. Persoanele care suferă de această boală poartă mutații în până la trei codoni ai genei PRNP.
În prionopatia cu susceptibilitate variabilă la protează, pacienții prezintă:
- tulburari cognitive
- severitatea extremă a tulburărilor psihiatrice: pot fi euforie și agitație, dar și apatie semnificativă
- disartrie
- afazie (tulburări ale funcțiilor limbajului)
Durata medie a bolii în această prionopatie este mai mică de 4 ani.
Encefalopatii spongiforme: kuru
Kuru este acum considerată o boală care practic nu mai există - a fost găsită la reprezentanții triburilor din Papua Noua Guinee, care practicau un comportament canibalist. Simptomul dominant al acestei encefalopatii spongiforme este ataxia cerebeloasă progresivă. Poate fi însoțit de mișcări involuntare (în principal sub formă de coree, tremor și atetoză), precum și incontinență urinară și fecală. Pacienții care suferă de kuru experimentează, de asemenea, modificări semnificative ale dispoziției, dezvoltând reflexe primitive (de exemplu, supt). O problemă destul de caracteristică în cazul acestei boli prionice sunt atacurile forțate de plâns sau râs - datorită acestui ultim fenomen, kuru este uneori denumit „moarte râzând”.
Encefalopatii spongiforme: diagnostic
Bolile prionice pot fi suspectate pe baza simptomelor pacientului. Cu toate acestea, acestea sunt destul de nespecifice, deoarece pot apărea și în cursul mai multor alte boli care nu sunt legate de prioni. Din acest motiv, în diagnosticul encefalopatiilor spongiforme sunt utilizate și următoarele:
- teste imagistice (de exemplu, imagistica prin rezonanță magnetică, care permite detectarea modificărilor legate de degenerarea creierului de către proteinele prionice),
- teste de laborator (cum ar fi evaluarea concentrațiilor de proteine în lichidul cefalorahidian, de exemplu proteine MAP-tau, S-100 sau 14-3-3),
- teste genetice (pentru a detecta prezența mutațiilor la pacient),
- teste imunhistochimice (folosirea anticorpilor împotriva proteinelor prionice).
Diagnosticul poate fi confirmat și prin autopsia creierului, în care este posibil să se găsească modificări caracteristice encefalopatiilor spongiforme. Acestea pot fi leziuni spongioase, distribuite diferit și cu o structură diferită (în funcție de entitatea specifică a bolii) plăci amiloide și defecte neuronale.
Encefalopatii spongiforme: tratament
Bolile prionice sunt în prezent incurabile - în ciuda numeroaselor studii care au loc de mulți ani, medicina încă nu are medicamente care ar putea încetini sau inhiba complet progresul lor. Tratamentul simptomatic este utilizat la pacienții cu encefalopatii spongiforme, care vizează ameliorarea intensității simptomelor și îmbunătățirea calității vieții lor cât mai mult posibil.
Cu toate acestea, se lucrează încă la tratamentul encefalopatiilor spongiforme. Oamenii de știință încearcă să folosească diverse metode - primul exemplu este terapia genică. Acestea ar afecta acizii nucleici și mutațiile prezente în structura lor - scopul aplicării terapiei genice ar fi neutralizarea erorilor din codul genetic. O abordare diferită stă la baza terapiei imune - se lucrează la crearea de anticorpi al căror rol ar fi eliminarea prionilor patogeni. O altă metodă care vede potențialul de a combate encefalopatiile spongiforme este tratamentul cu utilizarea moleculelor de proteine sintetizate, care, odată introduse în corpul pacientului, ar neutraliza proteinele patologice.
Articol recomandat:
Encefalopatii - Cauze, tipuri și simptome


-przyczyny-i-leczenie.jpg)
---choroba-objawiajca-si-blem-mini-i-koci.jpg)